
Introduction
Clinical Diagnosis of Prions is a critical aspect of modern medicine because these nontraditional infectious agents cause some of the rarest and most fatal neurodegenerative diseases in humans. Prions are abnormal proteins that lack genetic material and do not fit into the conventional categories of bacteria, viruses, or fungi. Unlike typical infectious agents, prions are acellular and trigger disease without causing the inflammation usually associated with infection. Prion diseases, also known as transmissible spongiform encephalopathies, progress rapidly and are universally fatal, making timely and accurate diagnosis essential. Understanding how to identify these unique agents through clinical methods allows healthcare professionals to differentiate prion diseases from other neurological disorders and to provide appropriate care focused on symptom management and patient comfort.

Overview of Prion Diseases
Definition and Alternate Name of Prion Diseases
Prion diseases, also known as transmissible spongiform encephalopathies or TSEs, are rare neurodegenerative disorders caused by abnormal proteins called prions. The study of prions is called prion biology and focuses on understanding what prions are, how they cause disease, and why they are so dangerous. Unlike viruses or bacteria, prions do not contain DNA or RNA, making them nontraditional infectious agents. These diseases are classified as nontraditional because they spread through protein misfolding rather than through conventional infection pathways, which makes clinical diagnosis of prions essential for detecting and managing these disorders.
Examples of Prion Diseases in Humans
Creutzfeldt-Jakob Disease (CJD)
Creutzfeldt-Jakob disease is the most common human prions disease. It can occur as a sporadic prion disease, inherited through genetic mutations, or as an acquired prion disease from exposure to contaminated tissue. Symptoms of CJD include rapid cognitive decline, loss of motor coordination, and personality changes. The disease progresses quickly, making early clinical diagnosis critical. CJD demonstrates why prions are so dangerous, as the misfolded proteins can induce other normal proteins to misfold, spreading the disease within the brain. Understanding CJD is essential in the study of prions and their impact on human health.
Variant Creutzfeldt-Jakob Disease (vCJD)
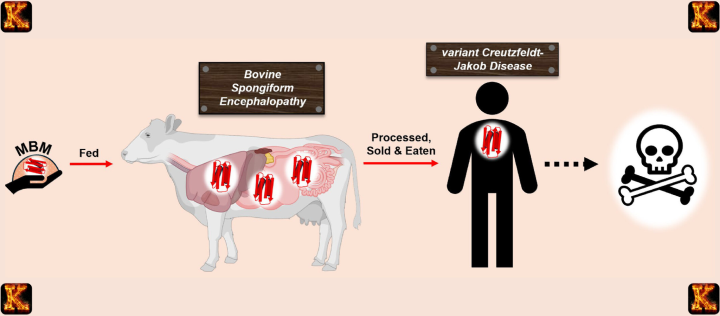
Variant CJD is an acquired prion disease linked to consuming meat infected with bovine spongiform encephalopathy. Patients often develop prion disease symptoms such as memory loss, behavioral changes, and movement difficulties. vCJD highlights how prions disease can be transmitted from animals to humans. This emphasizes the need for food safety and measures on how to avoid prion disease. Studying vCJD helps scientists understand what causes prion disease and why prions are so dangerous, as they remain resistant to conventional sterilization and can propagate without DNA or RNA.
Gerstmann-Sträussler-Scheinker Syndrome
Gerstmann-Sträussler-Scheinker syndrome is a hereditary prions disease caused by genetic mutations. Patients experience prion disease symptoms including progressive ataxia, memory impairment, and personality changes. The disease is inherited and demonstrates the role of genetics in prion disorders. Understanding this syndrome contributes to the study of prions and the mechanisms behind inherited prion disease. It also informs research on sporadic prion disease and acquired prion disease, expanding the list of prion diseases that affect humans and reinforcing the importance of recognizing early clinical symptoms.
Fatal Familial Insomnia
Fatal familial insomnia is another inherited prions disease that primarily affects sleep regulation. Patients experience severe insomnia, cognitive decline, and motor dysfunction. This prion disease demonstrates how misfolded proteins disrupt normal brain function. Studying this disease helps understand what prions are, why they are so dangerous, and how prion disease symptoms manifest differently depending on the affected brain region. Awareness of fatal familial insomnia also informs efforts on how to avoid prion disease exposure and expands the overall list of prion diseases known in humans.
Sporadic Prion Disease
Sporadic prion disease occurs without a clear cause and represents the majority of CJD cases. Patients develop prion disease symptoms such as rapid dementia, loss of coordination, and behavioral changes. Sporadic prion disease shows that not all prions disease are inherited or acquired. Studying sporadic cases is essential in understanding the mechanisms of prion propagation and how prions disease can arise spontaneously. It also contributes to the broader study of prions, the list of prion diseases, and highlights why prions are so dangerous despite the absence of direct infection or genetic inheritance.
Modes of Transmission of Prion Diseases
Acquired Prion Disease
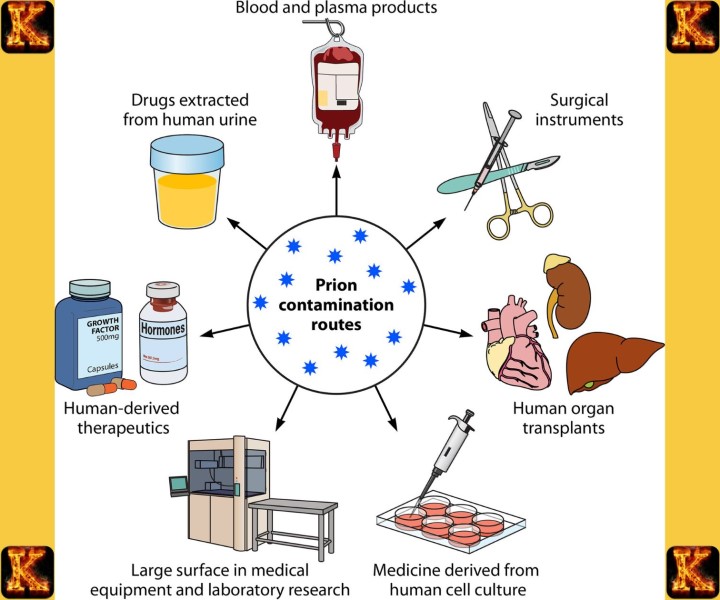
Acquired prion disease occurs when a person is exposed to infectious prion proteins from an external source. This form of prions disease can result from consuming contaminated meat, particularly meat linked to bovine spongiform encephalopathy, or through medical exposure such as contaminated surgical instruments, corneal transplants, or dura mater grafts. Understanding how prion disease is transmitted in acquired cases is essential for public health and infection control. Although prion disease is not contagious through casual contact, strict sterilization and screening procedures are necessary. Awareness of acquired prion disease helps explain why prions are so dangerous and why prevention strategies focus on reducing exposure risks.
Inherited Prion Disease
Inherited prion disease is caused by mutations in the gene that codes for the normal prion protein. These genetic changes lead to the production of misfolded proteins that trigger prion disease without external exposure. Examples from the list of prion diseases include fatal familial insomnia and Gerstmann-Sträussler-Scheinker syndrome. Inherited prions disease can be passed from parent to child, making family history an important diagnostic clue. Studying inherited cases contributes to the study of prions by helping researchers understand what causes prion disease at the molecular level. Genetic counseling is important for affected families, although there is currently no way to prevent disease onset.
Sporadic Prion Disease
Sporadic prion disease occurs spontaneously and accounts for the majority of human prion disease cases, particularly sporadic Creutzfeldt-Jakob disease. In these cases, prions form without a known trigger such as genetic mutation or exposure to infected tissue. This makes sporadic prion disease difficult to predict and challenging to prevent. The exact cause remains unclear, which is a major focus in the study of prions. Understanding sporadic cases helps clarify what prions are and how normal proteins can misfold on their own. Although prion disease is not contagious, sporadic cases highlight why prions are so dangerous and why early clinical recognition is critical.
Why Prions Are Dangerous
Unique Protein Structure and Self-Propagation
Prions are dangerous because they are composed solely of misfolded proteins that can force normal proteins in the brain to change shape. This abnormal folding process spreads without the need for DNA or RNA, which explains what prions are and why they behave differently from bacteria or viruses. Once misfolding begins, the damage amplifies rapidly throughout brain tissue. This mechanism is central to diseases caused by prions and explains what causes prion disease at the molecular level. The self-propagating nature of prions makes prions disease progressive, irreversible, and extremely difficult to control.
Resistance to Sterilization and Environmental Persistence
Another reason prions are so dangerous is their resistance to standard sterilization methods. Heat, radiation, and common disinfectants that kill bacteria and viruses are often ineffective against prions. This resistance increases the risk of acquired prion disease during medical procedures involving contaminated instruments or tissues. Understanding how prion disease is transmitted highlights why strict infection-control protocols are required in healthcare settings. Because prion disease is not contagious through casual contact, prevention focuses on minimizing exposure to infected material rather than isolation of patients.
Rapid and Fatal Disease Progression
Prions disease symptoms progress rapidly, leading to severe neurodegeneration and death within months to a few years. There are no treatments that can stop or reverse this process, making all prion diseases fatal. Rapid cognitive decline, motor dysfunction, and personality changes are common features across the list of prion diseases. This aggressive progression underscores why early recognition and clinical diagnosis are critical. Understanding why prions are so dangerous helps clinicians prioritize symptom management, patient comfort, and counseling for families affected by sporadic prion disease or acquired prion disease.
Challenges in Prevention and Treatment
Preventing prion disease is challenging because sporadic prion disease occurs without a known cause, and prions cannot be targeted by antibiotics or antivirals. Current prevention strategies focus on food safety, screening of donor tissues, and specialized sterilization procedures to reduce the risk of acquired prion disease. The study of prions continues to explore how to avoid prion disease and limit exposure risks. The absence of effective treatment reinforces the importance of understanding prions disease and highlights the need for continued research into these uniquely dangerous infectious agents.
Read Also: Effects of Probiotics: Impact on Health and Well-being
Common Signs and Symptoms of Prion Disease
Early Neurological and Cognitive Symptoms
Prion disease often begins with subtle neurological and cognitive changes. Individuals may experience difficulty concentrating, decreased attention span, or mild confusion. Early memory loss is common, affecting the ability to recall recent events while long-term memory remains intact initially. Behavioral changes, such as irritability, depression, or social withdrawal, may also appear. These early symptoms are often mistaken for normal aging or other neurological disorders. Recognizing these initial signs is important because prion diseases, including sporadic prion disease, progress rapidly, and early clinical observation can guide further diagnostic testing like cerebrospinal fluid analysis and EEG monitoring.
Memory Loss
Memory loss is a hallmark of prion disease symptoms and often appears early in the disease course. Patients may struggle to remember appointments, conversations, or personal items. As the disease progresses, short-term memory loss becomes more severe, contributing to rapid cognitive decline. This symptom is particularly significant in sporadic Creutzfeldt-Jakob disease, the most common prion disease in humans. Memory impairment is caused by the neurodegeneration and misfolding of proteins in critical areas of the brain, including the cortex and hippocampus. Understanding memory loss in prion disease helps differentiate it from other causes of dementia or neurodegenerative disorders.
Confusion
Confusion is another early symptom of prion diseases, including Creutzfeldt-Jakob disease. Affected individuals may become disoriented to time, place, or person and may struggle to make simple decisions. Confusion results from the progressive loss of normal brain function due to the accumulation of misfolded prion proteins. Because prion disease does not cause inflammation, these changes occur without typical signs of infection, making diagnosis challenging. Confusion often worsens rapidly over weeks to months, highlighting the aggressive nature of prion disease and the importance of clinical observation and diagnostic tests such as cerebrospinal fluid analysis and EEG patterns for early detection.
Behavioral Changes
Behavioral changes are common in prion disease patients. Mood swings, anxiety, depression, and uncharacteristic aggression may appear early in the disease. Some patients may exhibit apathy, social withdrawal, or unusual compulsive behaviors. These changes are caused by neurodegeneration in the frontal and temporal lobes of the brain. Recognizing behavioral alterations is important for distinguishing prion disease from other neurological or psychiatric conditions. In sporadic prion disease, these behavioral symptoms often accompany memory loss and confusion. Early awareness of such changes allows caregivers and clinicians to provide supportive care and monitor the disease’s rapid progression.
Lack of Inflammation
Unlike bacterial or viral infections, prion infections do not trigger an inflammatory response in the brain. This absence of inflammation makes prion disease difficult to detect in early stages using standard laboratory tests. The brain damage occurs silently, with misfolded prion proteins accumulating and causing neurodegeneration without the immune system responding. Understanding this feature helps explain why prion disease is often diagnosed late and why clinical diagnosis of prions relies on indirect markers such as elevated cerebrospinal fluid 14-3-3 protein levels and abnormal EEG patterns rather than traditional signs of infection.
Progressive Symptoms
Rapid Dementia
Rapid dementia is a defining feature of prion diseases. Unlike typical dementias that progress over years, cognitive decline in prion disease occurs over weeks to months. Patients lose the ability to reason, perform daily activities, and recognize loved ones. This accelerated deterioration is due to widespread neurodegeneration and the accumulation of misfolded prion proteins in multiple brain regions. Rapid dementia distinguishes prion diseases from more common neurodegenerative disorders like Alzheimer’s disease and is a critical diagnostic clue for clinicians performing the clinical diagnosis of prions.
Loss of Motor Coordination
Loss of motor coordination is another common progressive symptom of prion diseases. Patients may experience difficulty walking, frequent falls, tremors, or unsteady gait. Fine motor tasks, such as writing or buttoning clothes, become challenging as the disease advances. The underlying cause is the degeneration of neurons in the cerebellum and other motor control areas, which are vulnerable to the abnormal prion proteins. Monitoring motor coordination helps in identifying disease progression and differentiates prion disease from other neurological disorders that primarily affect cognition rather than movement.
Muscle Twitching
Muscle twitching, or myoclonus, is often observed in patients with prion diseases. These involuntary jerks can affect the limbs, face, or trunk and may occur at rest or during movement. Myoclonus results from abnormal electrical activity in the damaged brain regions caused by misfolded prion proteins. This symptom is particularly common in Creutzfeldt-Jakob disease and can help clinicians confirm the diagnosis alongside other tests such as EEG and cerebrospinal fluid analysis. Muscle twitching contributes to loss of motor function and overall physical decline in prion disease patients.
Example Focus: Sporadic Creutzfeldt-Jakob Disease
Sporadic Creutzfeldt-Jakob disease is the most common human prion disease, accounting for approximately 85% of cases. It occurs spontaneously, with no identifiable cause or exposure to infected tissue. Patients develop rapidly progressing neurological symptoms, including memory loss, confusion, behavioral changes, and motor dysfunction. Because sporadic prion disease is not caused by bacteria, viruses, or fungi, traditional treatments are ineffective. Diagnosis relies on clinical observation, EEG patterns, and elevated cerebrospinal fluid 14-3-3 protein levels. Awareness of sporadic CJD and its early signs is essential for providing supportive care and managing the rapid progression of this fatal prion disease.
Clinical Challenges in Diagnosing Prion Diseases
Why Prion Diseases Are Difficult to Diagnose
Prion diseases are challenging to diagnose because they progress rapidly and produce symptoms similar to other neurodegenerative disorders. Clinicians must differentiate sporadic prion disease from conditions like Alzheimer’s disease or Huntington’s disease. Traditional laboratory tests may not detect prion infections due to the acellular nature of prions. The study of prions emphasizes understanding what prions are and how they affect the nervous system. Clinical diagnosis often relies on specialized tests, including cerebrospinal fluid analysis for 14-3-3 protein and electroencephalogram patterns, rather than relying on standard infectious disease markers.
Symptoms Resemble Other Neurodegenerative Disorders
Prion disease symptoms can mimic other diseases caused by neurodegeneration, including memory loss, confusion, personality changes, and motor impairment. Rapid dementia and loss of coordination are common, making it difficult to recognize prions disease early. Because these symptoms overlap with more common disorders, clinicians may initially misdiagnose sporadic prion disease. Understanding prion disease symptoms and maintaining awareness of the full list of prion diseases is critical for clinical focus analyses. Early recognition is essential to guide palliative care and prevent further exposure when acquired prion disease is a possibility.
Lack of Inflammation or Immune Response
Unlike bacterial or viral infections, prion infections do not trigger an inflammatory response in the brain. This absence of immune activation makes it harder to detect prions using standard laboratory tests. Cerebrospinal fluid may show abnormal proteins such as 14-3-3, but no immune cells are present. The lack of inflammation explains why antibiotics and antivirals are ineffective. Clinicians rely on indirect markers and imaging studies to confirm a prion disease diagnosis. Understanding this feature helps students and healthcare workers learn how prion disease is transmitted and why prions are so dangerous compared to other infectious agents.
Why Antibiotics and Antivirals Are Ineffective
Prions are not bacteria or viruses and do not contain DNA or RNA, so treatments targeting conventional microbes are ineffective. Antibiotics cannot stop prions from inducing misfolding of normal proteins in the brain, and antiviral medications have no effect on prion replication. This limitation emphasizes the need for early clinical diagnosis of prions and symptomatic management. Clinicians focus on symptom relief and preventing complications in patients with sporadic prion disease or acquired prion disease. Understanding the unique biology of prions is central to why prions disease requires different diagnostic and therapeutic approaches from other infectious diseases.
Prions Are Not Bacteria or Viruses
Prions are nontraditional infectious agents that consist only of misfolded proteins and lack nucleic acids. They are extremely small and resistant to standard sterilization, making them difficult to control. Because prions are neither bacteria nor viruses, they are not listed on the tree of life and do not elicit an immune response. These characteristics make clinical diagnosis of prions particularly challenging. Awareness of what causes prion disease and how prion disease is transmitted helps healthcare workers advise patients on how to prevent prion disease and recognize early signs of prions disease symptoms.
Read Also: Role of the Nurse in Prevention of Cross Infection
Role of Cerebrospinal Fluid (CSF) Analysis
Purpose of CSF Testing in Suspected Prion Disease
Cerebrospinal fluid analysis is a critical clinical tool when evaluating patients for prions disease. CSF testing allows clinicians to detect abnormal proteins and biochemical markers that suggest neuronal injury. The study of prions focuses on understanding what prions are and how they cause neurodegeneration, and CSF testing provides evidence for these processes. In suspected cases of sporadic prion disease or acquired prion disease, CSF samples can help differentiate prions from other neurodegenerative disorders such as Alzheimer’s or Huntington’s disease. Early CSF analysis supports timely clinical diagnosis of prion disease symptoms.
Significance of 14-3-3 Protein
The 14-3-3 protein is a key biomarker measured in CSF when diagnosing prions disease. Elevated 14-3-3 levels indicate rapid neuronal damage, which is characteristic of many prion diseases. Measuring this protein supports the identification of sporadic prion disease and can guide clinical focus analyses. While it is not exclusive to prions, the presence of 14-3-3 protein alongside rapid cognitive decline, motor impairment, and other prion disease symptoms strengthens the likelihood that the patient is experiencing a disease caused by prions.
Indicator of Rapid Neuronal Damage
High 14-3-3 protein levels in CSF reflect ongoing neuronal injury caused by prions. This marker is particularly useful because prion disease progresses quickly and does not elicit an immune response. Detecting rapid neuronal damage helps differentiate prions disease from other slower-developing neurodegenerative disorders. Monitoring these changes in CSF provides insight into disease severity and progression. Understanding what causes prion disease, including sporadic or acquired prion disease, allows clinicians to interpret CSF results in context and assess how dangerous prions are to brain function.
Elevated Levels Support a Prion Disease Diagnosis
Elevated 14-3-3 protein in CSF provides supporting evidence for a prion disease diagnosis. Combined with clinical examination, EEG patterns, and other neuroimaging studies, CSF testing contributes to a comprehensive approach to identifying prions. Because prions are acellular and not bacteria or viruses, standard infection markers are ineffective. Therefore, measuring proteins like 14-3-3 allows clinicians to confirm that prion disease symptoms are consistent with diseases caused by prions. This biomarker is especially relevant for sporadic prion disease, which accounts for the majority of Creutzfeldt-Jakob disease cases.
Limitations of CSF Testing
CSF testing is not entirely specific to prions disease. Other neurological conditions can sometimes cause elevated 14-3-3 protein, making it necessary to use CSF results alongside EEG, imaging studies, and clinical history. Understanding how prion disease is transmitted and the list of prion diseases helps clinicians interpret CSF findings accurately. CSF testing is most effective when combined with other diagnostic tools to provide a complete assessment. Despite limitations, it remains an essential part of the clinical diagnosis of prions and a practical method to guide decisions on managing prion disease symptoms.
EEG Patterns in Prion Diseases
Use of Electroencephalogram (EEG) in Diagnosis
Electroencephalogram, or EEG, is a clinical tool used in the diagnosis of prions disease. EEG records electrical activity in the brain and provides insight into neurological function. Because prion disease symptoms often resemble those of other neurodegenerative disorders, EEG helps distinguish diseases caused by prions from conditions like Alzheimer’s or Huntington’s. In sporadic prion disease or acquired prion disease, abnormal EEG patterns can support the clinical diagnosis. Combining EEG findings with CSF analysis and other tests strengthens the accuracy of identifying prions disease and aids in understanding what prions are and how they damage neurons.
Typical EEG Findings in Prion Disease
Patients with prion disease often exhibit characteristic EEG findings that suggest severe brain dysfunction. Common observations include slowed background activity and abnormal wave patterns. These changes reflect the rapid neurodegeneration caused by prions. Monitoring EEG in patients with prion disease symptoms helps track disease progression and supports the clinical diagnosis, especially in sporadic prion disease where the cause is unknown. Recognizing these patterns is important for clinicians studying prions and provides a noninvasive method to assess how prion disease affects neuronal function in real time.
Abnormal Brain Wave Patterns
Abnormal brain wave patterns are a hallmark of prions disease on EEG. These patterns include diffuse slowing and disruption of normal rhythms, which indicate widespread neuronal damage. Such abnormalities correlate with clinical manifestations such as memory loss, confusion, and motor impairment. EEG abnormalities are not caused by infection or inflammation because prions do not trigger an immune response. Understanding these EEG signatures helps distinguish prion disease symptoms from other neurodegenerative disorders and contributes to studies on what causes prion disease and why prions are so dangerous.
Periodic Sharp Wave Complexes in Many CJD Cases
One of the most distinctive EEG findings in Creutzfeldt-Jakob disease is the presence of periodic sharp wave complexes. These patterns appear as repeated sharp waves occurring at regular intervals and are observed in many sporadic prion disease cases. Detecting these complexes is valuable for confirming the clinical diagnosis of prions disease, particularly when combined with CSF 14-3-3 protein levels. Periodic sharp wave complexes are not observed in most other neurodegenerative conditions, making this EEG feature a specific indicator of prions disease in humans.
How EEG Helps Distinguish Prion Disease from Other Conditions
EEG provides an important distinction between prions disease and other neurological disorders. While many diseases cause cognitive decline, few display the combination of diffuse slowing and periodic sharp wave complexes seen in prion disease. Using EEG alongside CSF testing and clinical assessment improves diagnostic confidence and guides clinicians in identifying diseases caused by prions. Understanding how prion disease is transmitted and recognizing sporadic prion disease cases further ensures accurate diagnosis. EEG serves as a practical tool to support the study of prions and the detection of prion disease symptoms early in disease progression.
Read Also: Per Patient Day System in Tackling Nurse Burnout
Why Prion Infections Lack Inflammation
Explanation of Immune Response Absence
Prion infections do not trigger a typical immune response because prions are acellular and lack DNA or RNA. Unlike bacteria or viruses, prions disease does not produce pathogen-associated molecular patterns that the immune system can recognize. As a result, the body does not mount an inflammatory reaction. This absence of immune activation contributes to the rapid progression of prions disease and the severity of prion disease symptoms. Understanding what prions are and why they are so dangerous highlights the unique challenges in diagnosing prion disease compared to other infectious neurological disorders.
Prions Do Not Trigger Typical Infection-Related Inflammation
Diseases caused by prions differ from viral or bacterial infections because they do not stimulate inflammation in the brain. Normally, infection triggers swelling, fever, or recruitment of white blood cells, but prion disease lacks these hallmarks. This lack of inflammation makes sporadic prion disease and acquired prion disease difficult to detect in early stages. Clinicians studying prions must rely on indirect indicators, such as CSF biomarkers and EEG patterns, to identify prion disease. Recognizing this feature is essential for distinguishing prion disease from other causes of encephalopathy.
No White Blood Cell Response in CSF
In prions disease, cerebrospinal fluid analysis shows no increase in white blood cells, which is a standard marker for infection. This absence indicates that prions disease is not caused by bacteria or viruses and helps rule out viral or bacterial encephalitis. The lack of white blood cell response, combined with elevated 14-3-3 protein levels, provides critical evidence in the clinical diagnosis of prion disease. Studying prions helps explain why the brain’s immune system does not respond and why prion disease symptoms progress unchecked, emphasizing why prions are so dangerous.
Clinical Importance
The absence of inflammation in prion disease has practical implications for diagnosis. It helps clinicians distinguish prions disease from other neurological infections that produce similar cognitive and motor symptoms. When combined with EEG findings and CSF testing, the lack of inflammatory response supports an accurate diagnosis of sporadic prion disease or acquired prion disease. This knowledge also informs public health strategies on how to avoid prion disease and emphasizes the importance of understanding what prions are and how prion disease is transmitted. Early recognition is critical since prion disease remains universally fatal.
Confirmatory Diagnosis
Brain Biopsy or Autopsy as Definitive Diagnosis
The most definitive method to diagnose prions disease is through examination of brain tissue. Brain biopsy or postmortem autopsy can directly identify the abnormal prion proteins and the characteristic sponge-like degeneration of neural tissue. This approach confirms what causes prion disease and distinguishes sporadic prion disease from acquired prion disease or inherited forms. Although effective, it is rarely performed during life due to the invasive nature of the procedure. Brain tissue analysis remains the gold standard and is essential for compiling an accurate list of prion diseases and understanding how prion disease is transmitted.
Ethical and Clinical Limitations of Confirmatory Testing During Life
Confirmatory diagnosis of prion disease through brain biopsy carries significant ethical and clinical challenges. Because prions disease is universally fatal and there is no approved treatment, invasive testing offers limited clinical benefit for the patient. Risk of surgical complications and potential exposure to infectious prion material makes biopsies ethically complex. Consequently, clinicians rely on noninvasive diagnostic tools such as CSF analysis, EEG, and clinical observation to support the diagnosis of prions disease. Understanding the study of prions highlights why confirmatory testing is rarely pursued during life and reinforces the need to prevent prion disease where possible.
Read Also: Nurses’ Role in Healthcare Capital Budgeting
Conclusion
Clinical diagnosis of prions relies on recognizing a combination of distinctive features. Elevated CSF 14-3-3 protein, abnormal EEG patterns, and the absence of inflammation in the brain provide strong evidence for prions disease. These indicators help distinguish prion infections from viral or bacterial encephalitis and other neurodegenerative disorders. Understanding what are prions and how prion disease symptoms present is critical for accurately diagnosing sporadic prion disease, acquired prion disease, and inherited forms. Clinicians must integrate these findings with patient history and progression of neurological symptoms to support a working diagnosis.
Importance of CSF 14-3-3 Protein and EEG Abnormalities
The presence of 14-3-3 protein in cerebrospinal fluid indicates rapid neuronal damage, a hallmark of prions disease. EEG abnormalities, including periodic sharp wave complexes, further support diagnosis by revealing specific brain wave patterns associated with prion disease. Together, these tools allow for clinical confirmation without invasive procedures. Identifying these markers helps understand how prion disease is transmitted, what causes prion disease, and why prions are so dangerous. Their use emphasizes the relevance of studying prions in microbiology and medicine to improve awareness of rare but fatal diseases caused by prions.
Absence of Inflammation
Prions disease is unique because it does not trigger a typical inflammatory response. No white blood cell infiltration occurs in the CSF, differentiating prion infections from viral or bacterial encephalitis. Recognizing this absence is essential for accurate clinical diagnosis and for advising patients on how to avoid prion disease exposure. The lack of inflammation underscores why prions are nontraditional infectious agents and why diagnosis relies on specialized tests rather than standard infection markers.
Importance of Clinical Pattern Recognition
Because prions disease is rare and universally fatal, clinical pattern recognition is critical. Physicians must correlate rapid cognitive decline, motor dysfunction, CSF biomarkers, EEG findings, and lack of inflammation to identify prions disease accurately. Awareness of prion disease symptoms supports early detection of sporadic prion disease or acquired prion disease. This clinical expertise is essential in the study of prions and in compiling a comprehensive list of prion diseases for research, epidemiology, and public health guidance on how to prevent prion disease.
Relevance in Microbiology and Medicine
Prion disease represents a unique intersection of microbiology, neurology, and infectious disease. Studying prions expands understanding of nontraditional infectious agents and their impact on human health. Accurate clinical diagnosis informs patient care, supports epidemiological tracking, and highlights the need for public awareness about how prion disease is transmitted. Recognizing prions disease contributes to the broader field of microbiology by emphasizing the importance of identifying diseases caused by prions and reinforces the need for safe practices to avoid prion disease in clinical and food-related settings.
Comments are closed!