Introduction
Tyrosinemia Type 1 is a rare and complex inherited metabolic disorder that affects individuals from infancy and poses significant challenges for both patients and their families. Characterized by the body's inability to properly break down the amino acid tyrosine, this condition leads to the accumulation of toxic substances that can cause severe damage to the liver, kidneys, and other organs. Due to its rarity, many healthcare providers and the general public lack awareness about the condition, making early diagnosis and access to appropriate care difficult. Understanding the underlying genetic causes, clinical manifestations, and treatment options is crucial for improving outcomes and quality of life for those affected. This introduction sets the stage for a detailed exploration of Tyrosinemia Type 1, highlighting the importance of raising awareness, promoting early detection, and providing comprehensive support to impacted individuals and their families.

Causes and Risk Factors
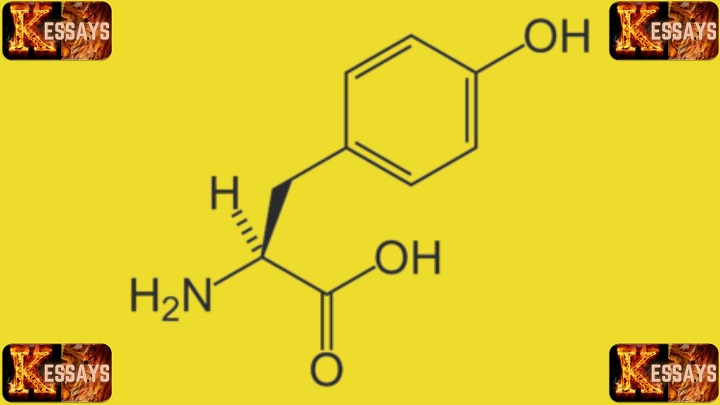
Tyrosinemia Type 1 is primarily caused by
genetic mutations in the
FAH gene, which encodes the enzyme fumarylacetoacetate hydrolase. This enzyme plays a crucial role in breaking down the amino acid tyrosine in the body. When both copies of the
FAH gene are mutated, individuals are unable to properly metabolize tyrosine, leading to the accumulation of toxic byproducts that can damage the liver, kidneys, and other organs.
Tyrosinemia Type 1 follows an autosomal recessive inheritance pattern. This means that an individual must inherit two defective copies of the
FAH gene, one from each parent, to develop the condition. Parents who carry only one faulty
FAH gene are typically asymptomatic but can pass the gene on to their children. If both parents are carriers, there is a 25% chance with each pregnancy that their child will inherit two defective copies of the gene and develop Tyrosinemia Type 1.
Several risk factors contribute to the likelihood of having a child with Tyrosinemia Type 1. The most significant is having both parents as carriers of the faulty
FAH gene. Individuals with a family history of Tyrosinemia Type 1 or who have previously had a child with the condition are at a higher risk. In some populations, genetic factors or consanguineous marriages (marriages between close relatives) may increase the prevalence of carriers. Additionally, new genetic mutations can sometimes arise spontaneously, resulting in Tyrosinemia Type 1 even in the absence of a family history.
Prenatal testing and genetic counseling are valuable tools for families at risk of Tyrosinemia Type 1. Genetic screening can identify carriers and assess the likelihood of passing the condition to future generations. By understanding the genetic basis and inheritance patterns of Tyrosinemia Type 1, families can make informed decisions about family planning and early intervention, ensuring timely diagnosis and management.
Signs and Symptoms of Tyrosinemia Type 1
Tyrosinemia Type 1 presents with a range of clinical features that can vary significantly in severity among affected individuals. Newborns with this condition may initially appear healthy but typically begin to show symptoms within the first few months of life.
Early Symptoms:
- Failure to thrive: Infants may struggle to gain weight and grow at a normal rate.
- Feeding difficulties: Many affected infants have trouble feeding, which can exacerbate poor weight gain.
- Vomiting and diarrhea: Gastrointestinal symptoms, including persistent vomiting and diarrhea, are common.
- Enlarged liver (hepatomegaly): The liver may become swollen, which can lead to abdominal distension.
Progressive Symptoms:
- Jaundice: As the disease progresses, affected infants may develop jaundice, characterized by yellowing of the skin and eyes.
- Neurological symptoms: These can include irritability, lethargy, and excessive sleepiness. In severe cases, neurological impairment may occur.
- Cabbage-like odor: A unique feature of Tyrosinemia Type 1 is a distinct, cabbage-like odor to the urine due to the accumulation of toxic substances.
Severe Complications:
- Liver failure: If untreated, Tyrosinemia Type 1 can cause progressive liver damage, ultimately leading to liver failure.
- Kidney dysfunction: Impaired kidney function can lead to issues such as proteinuria (excess protein in the urine) and renal tubular damage.
- Bone problems: Rickets or weakened bones may occur due to impaired vitamin D metabolism.
- Liver cancer (hepatocellular carcinoma): In some cases, untreated individuals may develop liver cancer, highlighting the importance of early diagnosis and treatment.
The onset and severity of these symptoms can vary. Some individuals may exhibit a milder form of the condition, while others may experience severe symptoms early in life. Early recognition and diagnosis are critical to ensure timely management and to prevent serious, long-term complications.
If any of these symptoms are observed in an infant or child, it is essential to seek immediate medical attention for further evaluation and testing. Early intervention can significantly improve outcomes for individuals with Tyrosinemia Type 1.
Diagnosing Tyrosinemia Type 1
Diagnosing Tyrosinemia Type 1 involves a combination of clinical evaluation, laboratory tests, and genetic analysis. Here's an overview of the process:
-
Clinical Evaluation:
- The initial step includes reviewing the individual’s medical history and performing a physical examination. Healthcare providers look for characteristic symptoms, such as failure to thrive, hepatomegaly (enlarged liver), jaundice, and the cabbage-like odor of the urine.
-
Laboratory Testing:
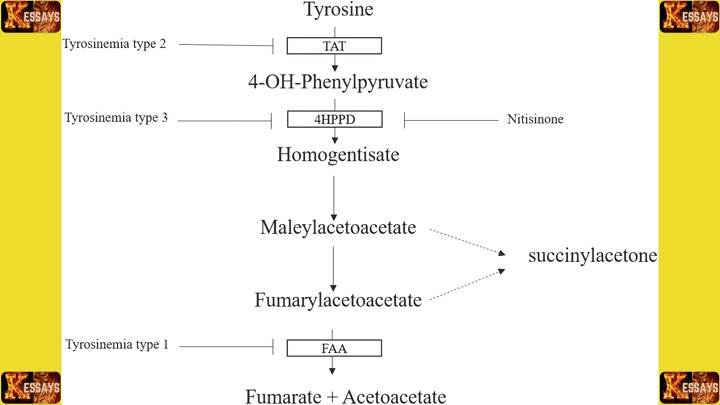
- Blood and Urine Tests: These are critical in detecting elevated levels of metabolites indicative of Tyrosinemia Type 1. Specifically, high levels of tyrosine and succinylacetone in the blood and urine are key diagnostic markers.
- Succinylacetone Testing: Measuring succinylacetone levels is one of the most reliable tests for diagnosing Tyrosinemia Type 1, as this metabolite accumulates due to the deficiency of the FAH enzyme.
-
Molecular Genetic Testing:
- Genetic testing is the gold standard for confirming a diagnosis. Testing can identify mutations in the FAH gene, which are responsible for Tyrosinemia Type 1.
- DNA Sequencing: This technique identifies specific mutations in the FAH gene.
- Gene Deletion/Duplication Analysis: It detects larger genetic alterations that may cause the condition.
- Targeted Mutation Analysis: In some cases, targeted analysis may be conducted based on common mutations seen in certain populations.
-
Newborn Screening:
- In countries where Tyrosinemia Type 1 is included in the newborn screening panel, the condition can be detected early, even before symptoms develop. This allows for immediate intervention, improving long-term outcomes.
-
Importance of Early Detection:
- Early detection is essential for prompt treatment. Interventions include dietary restrictions, particularly limiting the intake of tyrosine and phenylalanine, which helps prevent the buildup of toxic substances.
- Medication such as nitisinone (NTBC) is often prescribed early. Nitisinone blocks the production of harmful metabolites, including succinylacetone, helping to manage the condition and prevent complications.
-
Monitoring and Follow-up:
- Regular monitoring of blood and urine levels of tyrosine and succinylacetone is necessary to assess the effectiveness of treatment and make adjustments as needed.
-
Genetic Counseling:
- Genetic counseling is recommended for affected individuals and their families. It helps in understanding the inheritance pattern (autosomal recessive) and assessing the risk of having another child with Tyrosinemia Type 1 in future pregnancies. Prenatal genetic testing may also be offered.
In conclusion, accurate and early diagnosis of Tyrosinemia Type 1 is critical for initiating timely interventions. Proper dietary management, medication, and supportive care can significantly improve the quality of life for affected individuals, while minimizing the risk of long-term complications.
Treatment and Management Approaches for Tyrosinemia Type 1
Managing Tyrosinemia Type 1 involves a multidisciplinary approach aimed at reducing toxic metabolite accumulation, preventing organ damage, and improving long-term outcomes. The main treatment strategies include dietary modifications, medication, and in some cases, liver transplantation.
1. Dietary Modifications:
- Restriction of Tyrosine and Phenylalanine:
- A low-protein diet is a fundamental part of managing Tyrosinemia Type 1. This diet limits foods rich in tyrosine and phenylalanine, which are precursors to toxic by-products.
- A registered dietitian or nutritionist tailors the diet based on the individual’s specific needs, ensuring adequate nutrition while preventing excess accumulation of harmful metabolites.
- Specialized medical formulas and low-protein products are often necessary to supplement the diet, providing essential nutrients while keeping tyrosine and phenylalanine intake low.
- Regular monitoring of blood amino acid levels helps adjust the diet as needed to prevent deficiencies and maintain proper metabolic balance.
2. Medications:
- Nitisinone (Orfadin):
- Nitisinone is the primary medication used to manage Tyrosinemia Type 1. It works by inhibiting the enzyme that leads to the formation of toxic metabolites such as succinylacetone.
- The drug is usually prescribed alongside the low-protein diet to further prevent toxic accumulation.
- Ongoing monitoring of nitisinone levels in the blood and regular liver function tests are critical to ensure that the medication is working effectively and safely.
- Adjustments in the dosage may be needed based on the individual’s response to treatment.
3. Liver Transplantation:
- Indications for Transplant:
- In severe cases where dietary modifications and nitisinone fail to control the disease or where liver dysfunction has significantly advanced, liver transplantation may be considered.
- Surgical Replacement:
- A liver transplant replaces the diseased liver with a healthy donor liver that can break down tyrosine properly, halting the accumulation of toxic metabolites.
- Liver transplantation is considered curative because the new liver possesses the necessary enzyme (fumarylacetoacetate hydrolase, or FAH) to metabolize tyrosine normally.
- Post-Transplant Care:
- Following a transplant, lifelong immunosuppressive medications are required to prevent rejection of the new liver.
- Ongoing monitoring for complications is essential to ensure the transplanted liver functions well and to detect potential issues such as infections or organ rejection.
4. Collaborative Care and Long-term Management:
- Treatment of Tyrosinemia Type 1 involves a coordinated effort between metabolic specialists, dietitians, hepatologists, and transplant surgeons.
- Regular follow-ups are essential for:
- Monitoring of blood and urine parameters (such as tyrosine and succinylacetone levels).
- Adjustments to dietary plans and medications to optimize outcomes.
- Screening for complications, including liver damage, kidney dysfunction, and potential neurological issues.
5. Family Involvement and Support:
- It is important for individuals with Tyrosinemia Type 1 and their families to be actively involved in the management process.
- Education and support are critical for ensuring adherence to dietary restrictions, medication regimens, and follow-up care.
- Genetic counseling is also recommended for affected families to understand the inheritance pattern and assess risks for future pregnancies.
In conclusion, effective management of Tyrosinemia Type 1 requires a personalized and comprehensive approach, combining diet, medication, and, when necessary, liver transplantation. Early diagnosis, ongoing monitoring, and a strong partnership between healthcare providers and families are essential for achieving the best outcomes and enhancing the quality of life for affected individuals.
Complications and Long-Term Outlook
Tyrosinemia Type 1 can lead to various complications if not managed effectively. Understanding these potential complications is crucial for proactive intervention and long-term care. Additionally, considering the long-term prognosis and quality of life is essential for individuals and their families.
-
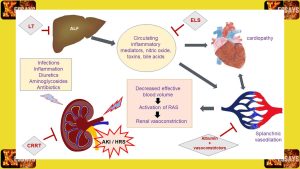
Liver Dysfunction and Failure:
- The accumulation of toxic metabolites in Tyrosinemia Type 1 can cause progressive liver damage and dysfunction.
- Without appropriate treatment, the liver may eventually fail, leading to complications such as jaundice, bleeding disorders, and fluid retention.
- Liver transplantation may be necessary to address advanced liver disease and prevent life-threatening complications.
-
Kidney Problems:
- Tyrosinemia Type 1 can affect kidney function, leading to the development of renal tubular dysfunction and renal Fanconi syndrome.
- Renal complications can manifest as electrolyte imbalances, urinary problems, and impaired kidney function.
-
Neurological Impairment:
- High levels of toxic metabolites in Tyrosinemia Type 1 can impact the central nervous system, leading to neurological symptoms and cognitive impairment.
- Neurological complications may include developmental delays, intellectual disabilities, seizures, and movement disorders.
-
Rickets and Bone Problems:
-
Increased Risk of Liver Cancer:
- Individuals with uncontrolled Tyrosinemia Type 1 have an increased risk of developing hepatocellular carcinoma, a type of liver cancer.
- Regular monitoring and appropriate management can help reduce this risk.
The long-term outlook for individuals with Tyrosinemia Type 1 largely depends on early detection, timely intervention, and adherence to treatment protocols. With proper management, including dietary modifications, medication, and potentially liver transplantation, the prognosis can be improved.
Regular monitoring of blood and urine parameters, liver function, and growth milestones is essential for assessing treatment efficacy and identifying any potential complications. Close collaboration with a multidisciplinary healthcare team, including metabolic specialists, hepatologists, and other relevant specialists, is crucial in providing comprehensive care and addressing potential complications as they arise.
Although Tyrosinemia Type 1 poses significant challenges, advancements in medical care, improved awareness, and support networks have contributed to enhanced quality of life for affected individuals. Early diagnosis, appropriate interventions, and ongoing care can greatly improve the long-term outlook and overall well-being of individuals with Tyrosinemia Type 1, allowing them to lead fulfilling lives to the best extent possible.
Support and Resources
Living with Tyrosinemia Type 1 can be challenging for individuals and their families. Having access to support networks, resources, and organizations dedicated to assisting those affected by the condition is crucial. These resources provide valuable information, guidance, and emotional support to navigate the complexities of managing Tyrosinemia Type 1.
-
Support Networks:
- Connecting with other individuals and families who have firsthand experience with Tyrosinemia Type 1 can be invaluable. Online forums, social media groups, and local support groups provide opportunities to share experiences, ask questions, and find encouragement.
- Local support groups can help facilitate connections with nearby families, enabling face-to-face interactions and the sharing of resources and knowledge.
-
Medical Professionals and Clinics:
- Establishing a strong relationship with healthcare professionals experienced in managing metabolic disorders is essential. These specialists, including metabolic physicians, dietitians, hepatologists, and genetic counselors, can provide comprehensive care, guidance, and ongoing support.
- Regular visits to specialized metabolic clinics allow for monitoring of treatment effectiveness, adjustments to dietary plans, and addressing any concerns or questions.
-
Patient and Family Education:
- Access to accurate and up-to-date information about Tyrosinemia Type 1 is crucial for individuals and their families. Educational materials, brochures, and online resources provided by reputable organizations can enhance understanding of the condition, treatment options, and potential challenges.
- Educational programs and workshops conducted by healthcare professionals and patient advocacy groups can offer in-depth information, practical tips, and opportunities for direct interaction and learning.
-
Advocacy and Research Organizations:
- Several organizations are dedicated to supporting individuals and families affected by Tyrosinemia Type 1. These organizations promote awareness, fund research initiatives, and offer resources to enhance the quality of life for those with the condition.
- Examples of such organizations include the American Association for Pediatric Liver Disease (AASLD), the National Organization for Rare Disorders (NORD), and the Tyrosinemia Society.
-
Financial and Social Support:
- Managing Tyrosinemia Type 1 may come with financial burdens due to the cost of medical care, specialized diets, and potential travel expenses for consultations or treatments. Exploring available financial support programs, grants, and insurance resources can alleviate some of these financial challenges.
- Social workers, patient advocates, and local community organizations may also offer assistance in navigating insurance, accessing government programs, and connecting individuals and families with other available resources.
Having access to support networks, resources, and organizations dedicated to Tyrosinemia Type 1 can empower individuals and families to better manage the condition and improve their overall well-being. The sharing of experiences, knowledge, and emotional support within these networks fosters a sense of community, reduces isolation, and provides hope and encouragement throughout the journey of living with Tyrosinemia Type 1.
Research and Future Directions
Ongoing research efforts in Tyrosinemia Type 1 are aimed at deepening our understanding of the condition, improving diagnostic methods, and developing more effective treatments. Advances in research contribute to enhancing the quality of life for individuals affected by Tyrosinemia Type 1. Here are some key areas of investigation and potential therapeutic interventions being explored:
-
Improved Diagnostic Techniques:
- Researchers are continuously working on refining and improving diagnostic methods for Tyrosinemia Type 1.
- Efforts are focused on developing more efficient and accessible screening tools, including advancements in newborn screening programs, to ensure early detection and prompt intervention.
-
Novel Therapeutic Approaches:
- Investigational therapies and new treatment modalities are being explored to address the underlying metabolic abnormalities in Tyrosinemia Type 1.
- Enzyme replacement therapy (ERT) is one approach under investigation, aiming to provide the deficient enzyme FAH to individuals with Tyrosinemia Type 1.
- Gene therapy, which involves introducing functional FAH genes into affected cells, holds promise as a potential curative approach.
- Small molecule drugs that target specific metabolic pathways and reduce the toxic metabolites are also being investigated.
-
Liver Support Systems:
- Advancements in liver support systems, such as bioartificial liver devices, are being explored as a potential bridge to transplantation or as a supportive measure in severe cases of Tyrosinemia Type 1.
- These systems aim to provide temporary liver function support, allowing time for liver transplantation or recovery.
-
Understanding Disease Progression:
- Research efforts are directed towards understanding the natural course and long-term outcomes of Tyrosinemia Type 1.
- Studying disease progression, the impact of varying treatment regimens, and the factors influencing treatment response provide valuable insights for optimizing management strategies.
-
Gene Editing Technologies:
- Emerging gene editing technologies, such as CRISPR-Cas9, hold promise for correcting the genetic mutations underlying Tyrosinemia Type 1.
- Preclinical studies using these technologies are ongoing to evaluate their potential for correcting FAH gene mutations and restoring normal enzyme function.
These research endeavors offer hope for future advancements in the diagnosis, treatment, and management of Tyrosinemia Type 1. As scientific understanding expands and innovative therapies emerge, the outlook for affected individuals may continue to improve, leading to better outcomes and an enhanced quality of life.
Collaboration among researchers, healthcare professionals, patient advocacy groups, and affected individuals and their families is crucial to advancing the field of Tyrosinemia Type 1 research. By supporting and participating in research initiatives, we can contribute to a better future for individuals living with this rare metabolic disorder.
Conclusion
In conclusion, Tyrosinemia Type 1 is a rare inherited metabolic disorder that affects individuals from an early age. Understanding its causes, recognizing its signs and symptoms, and diagnosing it promptly are crucial for effective management. With ongoing research and support from healthcare professionals, individuals affected by Tyrosinemia Type 1 can receive the necessary treatment and support to optimize their
quality of life. By raising awareness and supporting further research efforts, we can contribute to a brighter future for those living with Tyrosinemia Type 1 and their families.
Comments are closed!